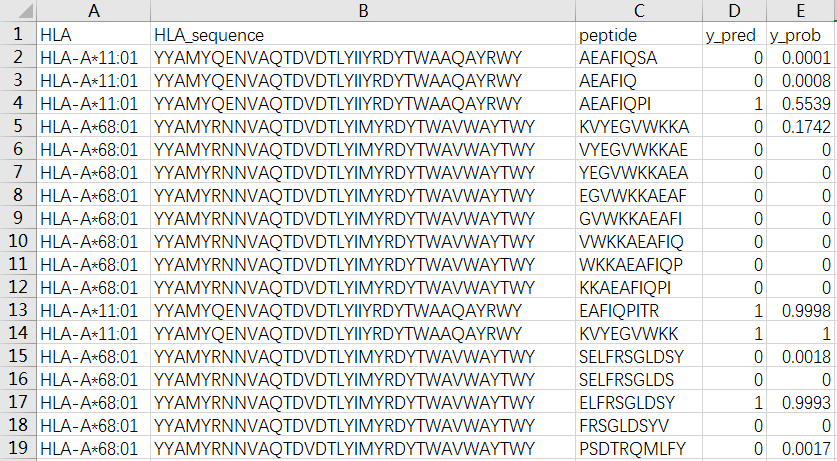
1. Input HLA and peptide sequences
➤ Although HLA and peptide are required to be entered separately, they will be combined into a peptide-HLA pair line by line. Therefore, the number of HLAs and peptides must be the same, otherwise, we will remind the user. Please be careful not to enter blank lines.
➤ The sequence must be provided in FASTA format. Whenever the ">" character appears, it means that an HLA or peptide appears, and will merge to obtain a peptide-HLA pair.
➤ All sequences must be amino acids specified with a single capital letter code (ACDEFGHIKLMNPQRSTVWY). If there are unknown letters or non-letters, the peptide-HLA pair will be filtered. If a lowercase letter appears, it will be automatically changed to an uppercase letter.
➤ We provide two sequence input methods: (1) Input directly in the list box, you can click the "Example" button to view the example. The example contains two HLAs. Correspondingly, two peptides should be entered in a one-to-one correspondence to form two peptide-HLA pairs. (2) Upload the FASTA file, you can click the "?" icon on the left to get an example. The example contains 5 HLAs. Correspondingly, 5 peptides should be entered to form 5 peptide-HLA pairs.
➤ If you enter the list box and upload the FASTA file at the same time, our choices are as follows:
◆ The list box is empty and the file is not uploaded: an error is reported
◆ The list box is not empty, and the file is not uploaded: use the list box
◆ The list box is empty, upload file: use file
◆ The list box is not empty, upload file: use file
➤ Limit on the number of sequences: None, but it is not recommended to exceed 5000.
➤ Length of sequence:
◆ For HLA: The sequence length requirement is ≤34. We provide 112 common HLA sequences. If you don’t know the HLA sequence, you can click on the "Some common HLA sequences" button to search.
◆ For peptide: The sequence length must be ≤15. However, since the TransPHLA-AOMP is trained on lengths of 8-14, other lengths may not achieve correct results. If the sequence length exceeds 15, we will cut according to the cut_length selected by the user, see Section 2 for details.
2. Split the input peptide sequence into multiple subsequences
➤ For peptide sequences greater than 15 in length, they will be forced to be cut into multiple peptide sub-sequences of "Cut length" selected by the user. We will discard the original peptide.
➤ If the user selects "Yes" on the question "Do you want to cut the sequence larger than cut_length you choose below?", the sequence with a length> "Cut length" will be cut into multiple sub-peptides with a length of "Cut length". Note that we will keep the original peptide.
➤ For the segmented multiple sub-peptides, we will pair them with the target HLA of the source peptide to form peptide-HLA pairs.
➤ Example of peptides with a sequence length > 15:
◆ Source peptide (sequence length is 17):
>KVYEGVWKKAEAFIQPI
KVYEGVWKKAEAFIQPI
◆ Target HLA:
>HLA-A*68:01
YYAMYRNNVAQTDVDTLYIMYRDYTWAVWAYTWY
◆ Cut length = 9
◆ The 9 peptides separated are:
KVYEGVWKK,VYEGVWKKA,YEGVWKKAE,EGVWKKAEA,GVWKKAEAF,VWKKAEAFI,WKKAEAFIQ,KKAEAFIQP,KAEAFIQPI
◆ The format after segmentation is:
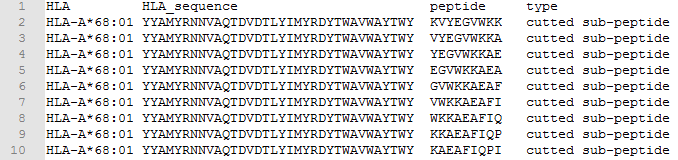
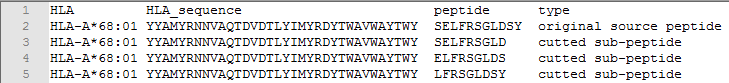
➤ Example of peptides with sequence length <15:
◆ Source peptide (sequence length is 11):
>SELFRSGLDSY
SELFRSGLDSY
◆ Target HLA:
>HLA-A*68:01
YYAMYRNNVAQTDVDTLYIMYRDYTWAVWAYTWY
◆ Cut length = 9
◆ The 3 peptides separated are:
SELFRSGLD,ELFRSGLDS,LFRSGLDSY
◆ The format after segmentation is:
3. Do you want to get attention scores?
If you select "Yes", a CSV file will be output for each peptide-HLA sample, the file name is HLA_peptide_attention.csv. The file will be located in the /attention folder. It reveals the influence of each amino acid in the peptide and HLA-I on whether it is bound. If there are N peptide-HLA samples, N corresponding CSV files will be generated.
Taking HLA-A*68:01 and peptide SDTRQMLFY as an example, the file will be saved as /attention/HLA-A*68:01_SDTRQMLFY_attention.csv, the details are as follows.
◆ Column A is the sequence of HLA-A*68:01.
◆ Row 1 is the sequence of the peptide.
◆ The element value is the attention score of the amino acid site of the peptide to the different amino acid sites of HLA. If the predicted result is binding, it is the contribution to the binding of peptide-HLA; if the predicted result is non-binding, it is the impact on the non-binding of peptide-HLA.
◆ Row "sum" indicates the sum of the attention score of the amino acid site on the peptide.
◆ Row "posi" indicates the position of the amino acid on the peptide.
◆ Row "contrib" indicates the contribution rate of the amino acid site of the peptide to whether or not the peptide-HLA is bound.

4. Do you want to save the images of attention scores?
◆ The heat map with DPI=600 is about 2MB in size for each sample, the file output is in the /figures folder, and the file name is HLA_peptide_predicted results_predicted score.tif.
◆ If there are N peptide-HLA samples, N corresponding tif files will be generated.
◆ The total number of samples must <1000 (including the samples generated by segmentation), otherwise even if the user selects "Yes", it will not be generated and output.
◆ The heatmap can be generated by attention.csv in Section 3. If the amount of data is too large, users can use the code provided by attention.py in GitHub to generate it.
◆ Taking HLA-A*68:01 and the peptide SDTRQMLFYf as an example, TransPHLAI predicts it as non-binding, and the predicted score (predicted as the probability of binding) is 0.0001, so the file name is HLA-A*68:01_SDTRQMLFY_Negative_Prob0.0001.tif. The brighter the color (yellow), the more important the site is to the prediction result.

5. Run the AOMP program to find higher affinity mutant peptides to the target HLA and high homology with original peptide?
AOMP is an automatically optimize mutated peptide program, it searches mutant peptides with higher affinity for the target HLA and the mutation between 1-4 positions according to the source peptide. This program not only guarantees the affinity between the mutant peptide and the target HLA, but also ensures the homology of the mutant peptide and the source peptide. With the source peptide, the homology of 1, 2, 3, and 4 amino acid positions were mutated on average at 90%, 80%, 70%, and 61%, respectively.
If you select "Yes", we will perform the AOMP program to find the high affinity mutated peptide to the target HLA, and output a CSV file for each peptide-HLA sample, the file name is HLA_peptide_mutation.csv. The file will be located in the /mutation folder.
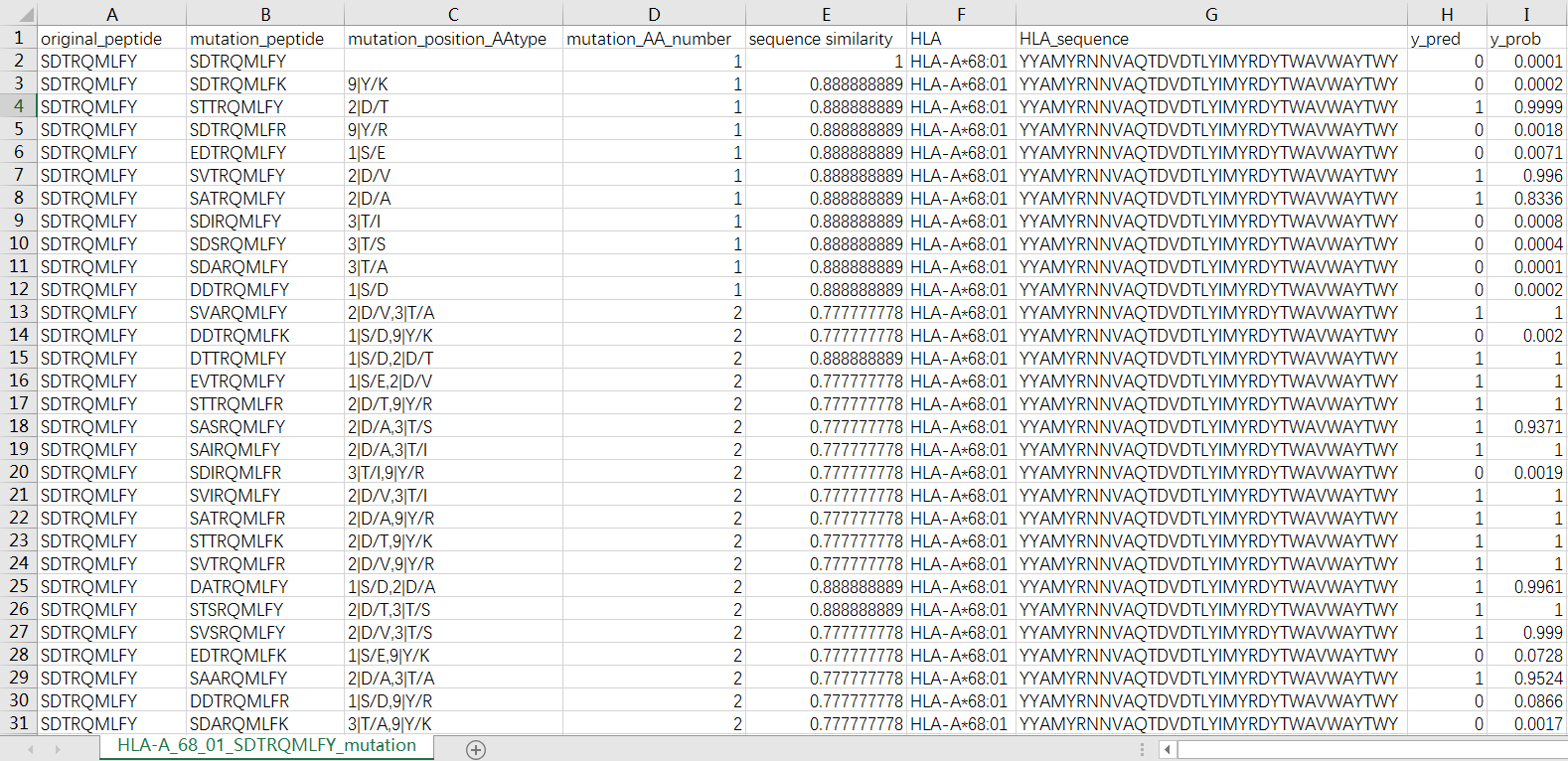
Taking HLA-A*68:01 and peptide SDTRQMLFY as an example, the file will be saved as /mutation/HLA-A*68:01_SDTRQMLFY_mutation.csv, the details are as follows.
6. The threshold to define predicted binder(0-1)?
We recommend setting it to 0.5, because TransPHLA is trained at 0.5.
7. The predict_results.csv
This file records the prediction results of peptide-HLA binding.